







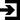
Scientific Applications:
NAMD - Molecular Dynamics
NAMD is the result of an interdisciplinary collaboration between Prof. Kale, computer science Prof. Robert D. Skeel, and physics Prof. Klaus J. Schulten at the Theoretical and Computational Biophysics Group (TCBG) of Beckman Institute.
NAMD is a parallel, object-oriented molecular dynamics code designed for high-performance simulation of large biomolecular systems. NAMD is distributed free of charge and includes source code. Charm++, developed by Prof. Kale and co-workers, simplifies parallel programming and provides automatic load balancing, which was crucial to the performance of NAMD.
In SC2002, Prof. Kale and co-authors James C. Phillips, Gengbin Zheng and Sameer Kumar, won a Gordon Bell Award for special accomplishment for their paper NAMD: Biomolecular Simulation on Thousands of Processors.
NAMD is implemented using the Converse runtime system, and the major components of NAMD are written in Charm++.
Converse provides machine-independent interface to all popular parallel computers as well as workstation clusters. Converse also implements a data-driven execution model, allowing parallel languages such as Charm++ to support the dynamic behavior of NAMD's chunk-based decomposition scheme.
The dynamic components of NAMD are implemented in the Charm++ parallel language. It is composed of collections of C++ objects, which communicate by remotely invoking methods on other objects. This supports the multi-partition decompositions in NAMD. Also data-driven execution adaptively overlaps communication and computation. Finally, NAMD benefits from Charm++'s load balancing framework to achieve unsurpassed parallel performance.
NAMD developers also extensively use the Projections, a performance visualization tool in Charm++, to diagnose and optimize the performance of NAMD.
The official NAMD home page is located at TCBG NAMD Research.
Outside of NAMD, we are experimenting with alternative ways to parallelize molecular dynamics computations.
NAMD is a parallel, object-oriented molecular dynamics code designed for high-performance simulation of large biomolecular systems. NAMD is distributed free of charge and includes source code. Charm++, developed by Prof. Kale and co-workers, simplifies parallel programming and provides automatic load balancing, which was crucial to the performance of NAMD.
In SC2002, Prof. Kale and co-authors James C. Phillips, Gengbin Zheng and Sameer Kumar, won a Gordon Bell Award for special accomplishment for their paper NAMD: Biomolecular Simulation on Thousands of Processors.
NAMD is implemented using the Converse runtime system, and the major components of NAMD are written in Charm++.
Converse provides machine-independent interface to all popular parallel computers as well as workstation clusters. Converse also implements a data-driven execution model, allowing parallel languages such as Charm++ to support the dynamic behavior of NAMD's chunk-based decomposition scheme.
The dynamic components of NAMD are implemented in the Charm++ parallel language. It is composed of collections of C++ objects, which communicate by remotely invoking methods on other objects. This supports the multi-partition decompositions in NAMD. Also data-driven execution adaptively overlaps communication and computation. Finally, NAMD benefits from Charm++'s load balancing framework to achieve unsurpassed parallel performance.
NAMD developers also extensively use the Projections, a performance visualization tool in Charm++, to diagnose and optimize the performance of NAMD.
The official NAMD home page is located at TCBG NAMD Research.
Outside of NAMD, we are experimenting with alternative ways to parallelize molecular dynamics computations.
People
Papers/Talks
20-03
2020
[Paper]
[Paper]
Scalable molecular dynamics on CPU and GPU architectures with NAMD [J. Chem. Phys. 2020]
16-19
2016
[Paper]
[Paper]
Handling Transient and Persistent Imbalance Together in Distributed and Shared Memory [PPL Technical Report 2016]
15-17
2015
[Talk]
[Talk]
Charm++ & MPI: Combining the Best of Both Worlds [IPDPS 2015]
15-02
2015
[Paper]
[Paper]
Charm++ & MPI: Combining the Best of Both Worlds [IPDPS 2015]
14-49
2014
[Talk]
[Talk]
Adaptive runtime systems for computational chemistry [ACS 2014]
13-43
2013
[Paper]
[Paper]
Parallel Science and Engineering Applications: The Charm++ Approach: Chapter 4: Scalable Molecular Dynamics with NAMD [Book 2013]
13-16
2013
[Paper]
[Paper]
Parallel Science and Engineering Applications: The Charm++ Approach [Book 2013]
12-55
2012
[Talk]
[Talk]
Fernbach Acceptance - NAMD [SC 2012]
12-33
2012
[Paper]
[Paper]
Optimizing Fine-grained Communication in a Biomolecular Simulation Application on Cray XK6 [SC 2012]
11-50
2012
[Paper]
[Paper]
A uGNI-Based Asynchronous Message-Driven Runtime System for Cray Supercomputers with Gemini Interconnect [IPDPS 2012]
11-17
2011
[Paper]
[Paper]
Enabling and Scaling Biomolecular Simulations of 100~Million Atoms on Petascale Machines with a Multicore-Optimized Message-Driven Runtime [SC 2011]
11-14
2011
[Poster]
[Poster]
Molecular Dynamics Simulations on Supercomputers Performing 10^18 flop/s [UIUC Postdoc Symposium 2011]
10-25
2011
[Paper]
[Paper]
NAnoscale Molecular Dynamics (NAMD) [Encyclopedia of Parallel Computing 2011]
10-20
2010
[Paper]
[Paper]
Periodic Hierarchical Load Balancing for Large Supercomputers [IJHPCA 2010]
10-15
2010
[Paper]
[Paper]
Simulating Large Scale Parallel Applications using Statistical Models for Sequential Execution Blocks [ICPADS 2010]
10-08
2010
[Paper]
[Paper]
Hierarchical Load Balancing for Charm++ Applications on Large Supercomputers [P2S2 2010]
09-15
2009
[Poster]
[Poster]
Performance Comparison of Intrepid, Jaguar and Ranger using Scientific Applications [SC 2009]
09-02
2009
[Paper]
[Paper]
Dynamic Topology Aware Load Balancing Algorithms for Molecular Dynamics Applications [ICS 2009]
08-01
2008
[Paper]
[Paper]
Overcoming Scaling Challenges in Biomolecular Simulations across Multiple Platforms [IPDPS 2008]
07-10
2007
[Paper]
[Paper]
NAMD: A Portable and Highly Scalable Program for Biomolecular Simulations [CS Res. & Tech. Report 2007]
07-05
2008
[Paper]
[Paper]
Biomolecular Modeling in the Era of Petascale Computing [Petascale Computing: Algorithms and Applications 2008]
07-02
2007
[Paper]
[Paper]
Scalable Molecular Dynamics with NAMD on Blue Gene/L [IBM Journal of Research and Development 2007]
06-20
2006
[Poster]
[Poster]
Charm++ on Cell [PPL Poster 2006]
06-19
2006
[Poster]
[Poster]
Charm++ Simplifies Programming for the Cell Processor [SC 2006]
05-26
2005
[Poster]
[Poster]
Speeding Up Parallel Simulation with Automatic Load Balancing [PPL Poster 2005]
05-13
2006
[Paper]
[Paper]
Achieving Strong Scaling with NAMD on Blue Gene/L [IPDPS 2006]
04-11
2004
[MS Thesis]
[MS Thesis]
LeanMD: A Charm++ Framework for High Performance Molecular Dynamics Simulation on Large Parallel Machines [Thesis 2004]
03-03
2003
[Paper]
[Paper]
Scaling Molecular Dynamics to 3000 Processors with Projections: A Performance Analysis Case Study [Workshop on Terascale Performance Analysis at ICCS 2003]
02-07
2002
[Paper]
[Paper]
NAMD: Biomolecular Simulation on Thousands of Processors [SC 2002]
02-04
2002
[Paper]
[Paper]
NAMD: Biomolecular Simulation on Thousands of Processors [Scaling to New Heights Workshop at Pittsburgh Supercomputing Center 2002]
00-06
2000
[Paper]
[Paper]
Scalable Molecular Dynamics for Large Biomolecular Systems [SC 2000]
98-10
1999
[Paper]
[Paper]
Algorithmic Challenges in Computational Molecular Biophysics [Journal of Computational Physics 1999]
98-08
1998
[Paper]
[Paper]
NAMD: A Case Study in Multilingual Parallel Programming [LCPC 1998]
98-06
1998
[Paper]
[Paper]
Computational Molecular Biophysics Today: A Confluence of Methodological Advances and Complex Biomolecular Applications [Journal of Computational Physics 1998]
98-05
1998
[Paper]
[Paper]
Avoiding Algorithmic Obfuscation in a Message-Driven Parallel MD Code [LNCS 1998]
98-04
1998
[Paper]
[Paper]
Flexibility and Interoperability in a Parallel Molecular Dynamics Code [Object Oriented Methods for Inter-operable Scientific and Engineering Computing 1998]
98-03
1998
[Paper]
[Paper]
NAMD2: Greater Scalability for Parallel Molecular Dynamics [Journal of Computational Physics 1998]
98-02
1998
[Paper]
[Paper]
Load Balancing in Parallel Molecular Dynamics [International Symposium on Solving Irregularly Structured Problems in Parallel 1998]
96-04
1996
[Paper]
[Paper]
NAMD - a Parallel, Object-Oriented Molecular Dynamics Program [International Journal Supercomputing Applications and High Performance Computing 1996]
94-02
1994
[Paper]
[Paper]
Modeling Biomolecules: Larger Scales, Longer Durations [IEEECSE 1994]